Stichproben-baseerte Quantendiagonalisierung vun een Chemie-Hamiltonian
Bruukschattung: ünner een Minuut op een Heron r2 Prozessor (HENWIES: Dat is blots een Schattung. Dien Looptiet kann variern.)
Achtergrund
In dit Tutorial wiesen wi, wo verrüschte Quantenstichproben naobearbeidt warrt, um een Approximation vun den Grundtostand vun dat Stickstoffmolekül bi Gliekgewichtsbindungslängde to finnen. Dobi bruukt wi den stichproben-baseerten Quantendiagonalisierungsalgorithmus (SQD) för Stichproben ut een 59-Qubit-Quantenschaltkreis (52 System-Qubits + 7 Ancilla-Qubits). Een Qiskit-baseerte Implementierung is verföögbar in de SQD Qiskit Addons, mehr Details finnen Se in de entsprechende Dokumentation mit een eenfach Bispeel to't Anfangen.
SQD is een Technik to't Finnen vun Eigenwerten un Eigenvektoren vun Quantenoperatoren, as den Hamiltonian vun een Quantensystem, ünner Bruuk vun Quanten- un verdeelte klassische Computing tosamen. Dat verdeelte klassische Computing warrt bruukt, um Stichproben vun een Quantenprozessor to verarbeiten un een Teel-Hamiltonian in een dör ehr opspannte Ünnerruum to projezeren un to diagonaliseren. Dat maakt SQD robust gegen Stichproben, de dör Quantenrüschen verfälscht sünd, un to't Ümgahn mit grote Hamiltonians, as Chemie-Hamiltonians mit Millionen vun Wechselwirkungstermen, buten de Riekwiedte vun exakte Diagonaliseringsmethoden. Een SQD-baseerte Workflow hett folgend Schritten:
- Wähl een Schaltkreis-Ansatz un wend em op een Quantencomputer op een Referenztostand an (in dit Fall den Hartree-Fock-Tostand).
- Sample Bitstrings ut den resulterende Quantentostand.
- Föhr dat sülvkonsistente Konfigurationswedderherstellenverfahren op de Bitstrings ut, um de Approximation vun den Grundtostand to kriegen.
SQD is bekannt dorför, goot to arbeiten, wenn de Teel-Eigentostand dünn besett is: De Wellenfunktion warrt dregen vun een Menge vun Basistoständ , ehr Grött nich exponentiell mit de Problemgrött wussen deit.
Quantenchemie
De Eigenschaften vun Molekülen warrt grootdeels dör de Struktur vun de Elektronen in ehr bestimmt. As fermionische Deelken künnt Elektronen mit een mathematischen Formalismus, Tweedquantisierung nömmt, beschreven warrn. De Idee is, dat dat een Antall vun Orbitalen gifft, vun de jeedeen entweder leer is or vun een Fermion besett warrt. Een System vun Orbitalen warrt dör een Satz fermionischer Vernichtungsoperatoren beschreven, de de fermionischen Antikommutatorrelatschonen erfüllen:
De adjungeerde Operator warrt Erzeugungsoperator nömmt.
Bit nu hett uns Darstellen de Spin nich berücksichtigt, de een fundamentale Eigenschaft vun Fermionen is. Bi't Berücksichtigen vun Spin kamen de Orbitalen in Poren vör, Ruumorbitalen nömmt. Jeed Ruumorbital besteiht ut twee Spinorbitalen, vun de een mit "Spin-" un een mit "Spin-" tekent warrt. Wi schrieven denn för den Vernichtungsoperator, de mit dat Spinorbital mit Spin () in Ruumorbital verbunnen is. Wenn wi as Antall vun de Ruumorbitalen nehmen, gifft dat insgesamt Spinorbitalen. De Hilbert-Ruum vun dit System warrt vun orthonormale Basisvektoren opspannt, de mit tweedelige Bitstrings tekent warrn: .
De Hamiltonian vun een molekular System kann schreven warrn as
wobei de un komplexe Tahlen sünd, de molekulare Integrale nömmt warrn un ut de Spezifikation vun dat Molekül mit een Computerprogramm berekent warrn künnt. In dit Tutorial berekent wi de Integrale mit dat Softwarepaket PySCF.
För Details dorüm, wo de molekulare Hamiltonian herleidt warrt, konsulteer een Lehrbök to Quantenchemie (to'n Bispeel Modern Quantum Chemistry vun Szabo un Ostlund). För een högerordnete Verklaren, wo Quantenchemieprobleme op Quantencomputer afbildt warrn, kiek di de Vorlesung Mapping Problems to Qubits vun de Qiskit Global Summer School 2024 an.
Local Unitary Cluster Jastrow (LUCJ) Ansatz
SQD bruukt een Quantenschaltkreis-Ansatz, ut den Stichproben trocken warrn künnt. In dit Tutorial bruukt wi den Local Unitary Cluster Jastrow (LUCJ) Ansatz wegen sien Kombination ut physikalische Motivation un Hardware-Fründlichkeit.
De LUCJ-Ansatz is een spezialiseerde Form vun den allgemenen Unitary Cluster Jastrow (UCJ) Ansatz, de de Form hett
wobei een Referenztostand is, faken de Hartree-Fock-Tostand, un de un de Form hebbt
wobei wi den Deelkentall-Operator defineert hebbt
De Operator is een Orbitalrotation, de mit bekannte Algorithmen in linearer Deepte un ünner Bruuk vun linearer Konnektivität implementeert warrn kann.
De Implementierung vun den Term vun den UCJ-Ansatz fordert entweder vollständige Konnektivität or den Bruuk vun een fermionisch Swap-Nettwark, wat för verrüschte prä-fehlertolerante Quantenprozessoren mit begrenzte Konnektivität een Roperfordderung is. De Idee vun den lokalen UCJ-Ansatz is, Dünnbessettheitsbedingen op de - un -Matrizen opto leggen, de dat möglich maken, ehr in konstante Deepte op Qubit-Topologien mit begrenzte Konnektivität to implementeren. De Bedingen warrn dör een List vun Indizes spezifizieret, de anwiesen, welk Matrixindrääg in dat bövere Drieheck vun null verscheden sien dröfft (do de Matrizen symmetrisch sünd, mutt blots dat bövere Drieheck spezifizieret warrn). Disse Indizes künnt as Orbitalpaore interpratseert warrn, de mitanner interageren dröfft.
Betrach as Bispeel een quadratisch Gitter-Qubit-Topologie. Wi künnt de - un -Orbitalen in parallele Lienen op dat Gitter platzeren, wobei Verbinnen twischen de Lienen "Sprossen" vun een Leiterforrm bilden, as folgt:

Bi dit Setup sünd Orbitalen mit den sülven Spin mit een Linientopologie verbunnen, wiel Orbitalen mit ünnersch Spin verbunnen sünd, wenn se dat sülve Ruumorbital delen. Dat gifft de folgend Indexbedingen för de -Matrizen:
Mit anner Wöör: Wenn de -Matrizen blots an de angeven Indizes in dat bövere Drieheck vun null verscheden sünd, kann de Term op een quadratische Topologie ahn Bruuk vun Swap-Gates in konstante Deepte implementeert warrn. Natüürlich maakt dat Opleggen vun söke Bedingen op den Ansatz em weniger utdrucksstark, sodass villicht mehr Ansatz-Wedderholen nöödig sünd.
De IBM® Hardware hett een Heavy-Hex-Gitter-Qubit-Topologie, in welk Fall wi een "Zickzack"-Muster bruken künnt, dat ünnen darstallt is. In dit Muster warrn Orbitalen mit den sülven Spin op Qubits mit een Linientopologie afbildt (roode un blaue Kringe), un een Verbinnen twischen Orbitalen vun ünnersch Spins is an jeeden 4. Ruumorbital vörhannen, wobei de Verbinnen dör een Ancilla-Qubit (violette Kringe) möglich maakt warrt. In dit Fall sünd de Indexbedingen
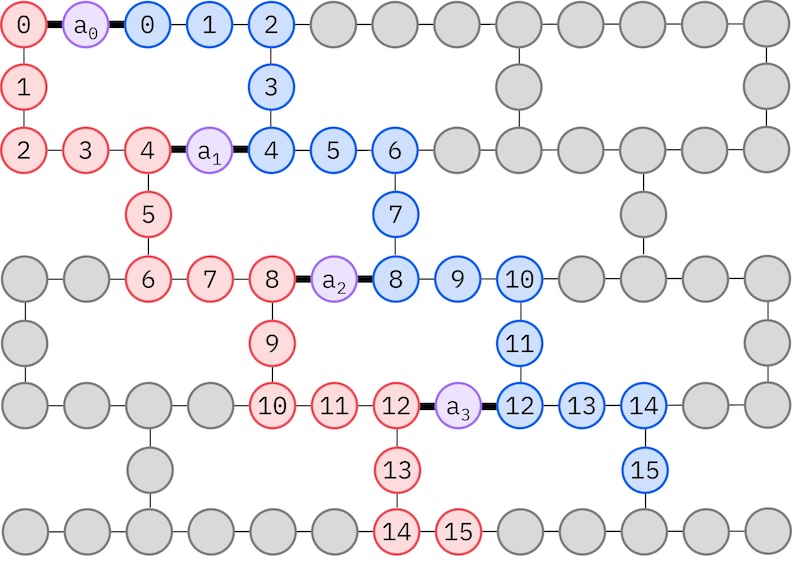
Sülvkonsistente Konfigurationswedderherstellung
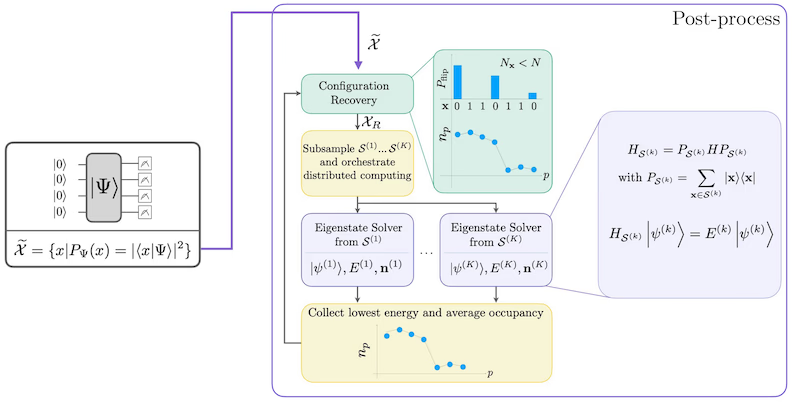
Dat sülvkonsistente Konfigurationswedderherstellenverfahren is dorop utleegt, so veel Signal as möglich ut verrüschte Quantenstichproben ruttohalen. Do de molekulare Hamiltonian de Deelkentall un Spin-Z erholen deit, maakt dat Sinn, een Schaltkreis-Ansatz to wählen, de disse Symmetrien ok erholen deit. Wenn he op den Hartree-Fock-Tostand anwendt warrt, hett de resulterende Tostand in den rüschenfrijen Fall een faste Deelkentall un Spin-Z. Dorher schullen de Spin-- un Spin--Hälvten vun jeeden ut den Tostand sampelte Bitstring dat sülve Hamming-Gewicht as in den Hartree-Fock-Tostand hebben. Wegen dat Vörhannen sien vun Rüschen in aktuelle Quantenprozessoren warrn welk mäten Bitstrings disse Eigenschaft verletzen. Een eenfache Forrm vun Postselektion würr disse Bitstrings wegsmiten, aver dat is verspillerisch, wiel disse Bitstrings villicht noch een beten Signal enthalden. Dat sülvkonsistente Wedderherstellenverfahren versöcht, een Deel vun dat Signal in de Naobearbeiten wedder hertostellen. Dat Verfahren is iterativ un bruukt as Ingaav een Schattung vun de dörchsnittlichen Bessetten vun jeeden Orbital in den Grundtostand, de toeerst ut de Rohstichproben berekent warrt. Dat Verfahren warrt in een Slööp utföhrt, un jeed Iteration hett de folgend Schritten:
- För jeeden Bitstring, de de spezifizierten Symmetrien verletzt, flipp sien Bits mit een probabilistisch Verfahren, dat dorop utleegt is, den Bitstring neger an de aktuelle Schattung vun de dörchsnittlichen Orbitalbessetten to bringen, um een nijen Bitstring to kriegen.
- Sammel all olen un nijen Bitstrings, de de Symmetrien erfüllen, un trock Deelmengen vun een in't Vörus wählte faste Grött.
- För jeed Deelmenge vun Bitstrings projezeer den Hamiltonian in den dör de entsprechend Basisvektoren opspannte Ünnerruum (kiek in den vörherigen Afsnitt för een Beschrieven vun de Basisvektoren) un berekne een Grundtostandsschattung vun den projezeerten Hamiltonian op een klassischen Computer.
- Aktualiseer de Schattung vun de dörchsnittlichen Orbitalbessetten mit de Grundtostandsschattung mit de neddrigste Energie.
SQD-Workflow-Diagramm
De SQD-Workflow is in dat folgend Diagramm darstallt:
Anfordderungen
Stell seker vör't Anfangen vun dit Tutorial, dat du Folgendes installeert hest:
- Qiskit SDK v1.0 or höger, mit Visualisierens-Ünnerst Süttung
- Qiskit Runtime v0.22 or höger (
pip install qiskit-ibm-runtime) - SQD Qiskit Addon v0.11 or höger (
pip install qiskit-addon-sqd) - ffsim v0.0.58 or höger (
pip install ffsim)
Setup
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime rustworkx
import pyscf
import pyscf.cc
import pyscf.mcscf
import ffsim
import numpy as np
import matplotlib.pyplot as plt
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
from qiskit_ibm_runtime import QiskitRuntimeService
from qiskit_ibm_runtime import SamplerV2 as Sampler
Schritt 1: Klassische Ingaav op een Quantenproblem afbilden
In dit Tutorial finnen wi een Approximation vun den Grundtostand vun dat Molekül in't Gliekgewicht in den cc-pVDZ-Basisatz. Toeerst spezifizeren wi dat Molekül un sien Eigenschaften.
# Specify molecule properties
open_shell = False
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="cc-pvdz",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
num_orbitals = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
num_elec_a = (n_electrons + mol.spin) // 2
num_elec_b = (n_electrons - mol.spin) // 2
cas = pyscf.mcscf.CASCI(scf, num_orbitals, (num_elec_a, num_elec_b))
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), num_orbitals)
# Store reference energy from SCI calculation performed separately
exact_energy = -109.22690201485733
converged SCF energy = -108.929838385609
Vör't Konstrueren vun den LUCJ-Ansatz-Schaltkreis föhrt wi toeerst een CCSD-Bereknung in de folgend Code-Zell dör. De - un -Amplituden ut de Bereknung warrn bruukt, um de Parameters vun den Ansatz to initialiseren.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -109.2177884185543 E_corr = -0.2879500329450045
Nu bruukt wi ffsim, um den Ansatz-Schaltkreis to maken. Do uns Molekül een slaten-schalich Hartree-Fock-Tostand hett, bruukt wi de spin-balanzierte Variante vun den UCJ-Ansatz, UCJOpSpinBalanced. Wi geven Wechselwirkungspaore, de för een Heavy-Hex-Gitter-Qubit-Topologie passt (kiek in den Achtergrundafsnitt to'n LUCJ-Ansatz). Wi setten optimize=True in de from_t_amplitudes-Methode, um de "komprimierte" Doppelfaktorisierung vun de -Amplituden to möglich maken (kiek The local unitary cluster Jastrow (LUCJ) ansatz in de ffsim-Dokumentation för Details).
n_reps = 1
alpha_alpha_indices = [(p, p + 1) for p in range(num_orbitals - 1)]
alpha_beta_indices = [(p, p) for p in range(0, num_orbitals, 4)]
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(alpha_alpha_indices, alpha_beta_indices),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell from running
# too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
nelec = (num_elec_a, num_elec_b)
# create an empty quantum circuit
qubits = QuantumRegister(2 * num_orbitals, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(num_orbitals, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Schritt 2: Problem för Quantenhardware-Utföhren optimeren
As Nächstes optimeren wi den Schaltkreis för een Teel-Hardware.
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=133
)
print(f"Using backend {backend.name}")
Using backend ibm_fez
Wi empfehlen de folgend Schritten, um den Ansatz to optimeren un hardware-kompatibel to maken.
- Wähl physikalische Qubits (
initial_layout) ut de Teel-Hardware ut, de to dat vörher beschreven "Zickzack"-Muster passt. Dat Anleggen vun Qubits in dit Muster föhrt to een effizienten hardware-kompatibeln Schaltkreis mit weniger Gates. Hier fögen wi Code in, um de Utwahl vun dat "Zickzack"-Muster to automatiseren, de een Bewertensh euristik bruukt, um de mit dat utwählte Layout verbunnen Fehler to minimeren. - Genereer een stufte Pass-Manager mit de Funktion generate_preset_pass_manager vun Qiskit mit dien Wahl vun
backenduninitial_layout. - Sett de
pre_init-Stuuv vun dien stufte Pass-Manager opffsim.qiskit.PRE_INIT.ffsim.qiskit.PRE_INITenthöllt Qiskit-Transpiler-Passes, de Gates in Orbitalrotationen updeelt un denn de Orbitalrotationen tosamanföhrt, wat to weniger Gates in den enngültigen Schaltkreis föhrt. - Föhr den Pass-Manager op dien Schaltkreis ut.
Code för automatiseerde Utwahl vun dat "Zickzack"-Layout
from typing import Sequence
import rustworkx
from qiskit.providers import BackendV2
from rustworkx import NoEdgeBetweenNodes, PyGraph
IBM_TWO_Q_GATES = {"cx", "ecr", "cz"}
def create_linear_chains(num_orbitals: int) -> PyGraph:
"""In zig-zag layout, there are two linear chains (with connecting qubits between
the chains). This function creates those two linear chains: a rustworkx PyGraph
with two disconnected linear chains. Each chain contains `num_orbitals` number
of nodes, that is, in the final graph there are `2 * num_orbitals` number of nodes.
Args:
num_orbitals (int): Number orbitals or nodes in each linear chain. They are
also known as alpha-alpha interaction qubits.
Returns:
A rustworkx.PyGraph with two disconnected linear chains each with `num_orbitals`
number of nodes.
"""
G = rustworkx.PyGraph()
for n in range(num_orbitals):
G.add_node(n)
for n in range(num_orbitals - 1):
G.add_edge(n, n + 1, None)
for n in range(num_orbitals, 2 * num_orbitals):
G.add_node(n)
for n in range(num_orbitals, 2 * num_orbitals - 1):
G.add_edge(n, n + 1, None)
return G
def create_lucj_zigzag_layout(
num_orbitals: int, backend_coupling_graph: PyGraph
) -> tuple[PyGraph, int]:
"""This function creates the complete zigzag graph that 'can be mapped' to an IBM QPU with
heavy-hex connectivity (the zigzag must be an isomorphic sub-graph to the QPU/backend
coupling graph for it to be mapped).
The zigzag pattern includes both linear chains (alpha-alpha interactions) and connecting
qubits between the linear chains (alpha-beta interactions).
Args:
num_orbitals (int): Number of orbitals, that is, number of nodes in each alpha-alpha linear chain.
backend_coupling_graph (PyGraph): The coupling graph of the backend on which the LUCJ ansatz
will be mapped and run. This function takes the coupling graph as a undirected
`rustworkx.PyGraph` where there is only one 'undirected' edge between two nodes,
that is, qubits. Usually, the coupling graph of a IBM backend is directed (for example, Eagle devices
such as ibm_brisbane) or may have two edges between two nodes (for example, Heron `ibm_torino`).
A user needs to be make such graphs undirected and/or remove duplicate edges to make them
compatible with this function.
Returns:
G_new (PyGraph): The graph with IBM backend compliant zigzag pattern.
num_alpha_beta_qubits (int): Number of connecting qubits between the linear chains
in the zigzag pattern. While we want as many connecting (alpha-beta) qubits between
the linear (alpha-alpha) chains, we cannot accommodate all due to qubit and connectivity
constraints of backends. This is the maximum number of connecting qubits the zigzag pattern
can have while being backend compliant (that is, isomorphic to backend coupling graph).
"""
isomorphic = False
G = create_linear_chains(num_orbitals=num_orbitals)
num_iters = num_orbitals
while not isomorphic:
G_new = G.copy()
num_alpha_beta_qubits = 0
for n in range(num_iters):
if n % 4 == 0:
new_node = 2 * num_orbitals + num_alpha_beta_qubits
G_new.add_node(new_node)
G_new.add_edge(n, new_node, None)
G_new.add_edge(new_node, n + num_orbitals, None)
num_alpha_beta_qubits = num_alpha_beta_qubits + 1
isomorphic = rustworkx.is_subgraph_isomorphic(
backend_coupling_graph, G_new
)
num_iters -= 1
return G_new, num_alpha_beta_qubits
def lightweight_layout_error_scoring(
backend: BackendV2,
virtual_edges: Sequence[Sequence[int]],
physical_layouts: Sequence[int],
two_q_gate_name: str,
) -> list[list[list[int], float]]:
"""Lightweight and heuristic function to score isomorphic layouts. There can be many zigzag patterns,
each with different set of physical qubits, that can be mapped to a backend. Some of them may
include less noise qubits and couplings than others. This function computes a simple error score
for each such layout. It sums up 2Q gate error for all couplings in the zigzag pattern (layout) and
measurement of errors of physical qubits in the layout to compute the error score.
Note:
This lightweight scoring can be refined using concepts such as mapomatic.
Args:
backend (BackendV2): A backend.
virtual_edges (Sequence[Sequence[int]]): Edges in the device compliant zigzag pattern where
nodes are numbered from 0 to (2 * num_orbitals + num_alpha_beta_qubits).
physical_layouts (Sequence[int]): All physical layouts of the zigzag pattern that are isomorphic
to each other and to the larger backend coupling map.
two_q_gate_name (str): The name of the two-qubit gate of the backend. The name is used for fetching
two-qubit gate error from backend properties.
Returns:
scores (list): A list of lists where each sublist contains two items. First item is the layout, and
second item is a float representing error score of the layout. The layouts in the `scores` are
sorted in the ascending order of error score.
"""
props = backend.properties()
scores = []
for layout in physical_layouts:
total_2q_error = 0
for edge in virtual_edges:
physical_edge = (layout[edge[0]], layout[edge[1]])
try:
ge = props.gate_error(two_q_gate_name, physical_edge)
except Exception:
ge = props.gate_error(two_q_gate_name, physical_edge[::-1])
total_2q_error += ge
total_measurement_error = 0
for qubit in layout:
meas_error = props.readout_error(qubit)
total_measurement_error += meas_error
scores.append([layout, total_2q_error + total_measurement_error])
return sorted(scores, key=lambda x: x[1])
def _make_backend_cmap_pygraph(backend: BackendV2) -> PyGraph:
graph = backend.coupling_map.graph
if not graph.is_symmetric():
graph.make_symmetric()
backend_coupling_graph = graph.to_undirected()
edge_list = backend_coupling_graph.edge_list()
removed_edge = []
for edge in edge_list:
if set(edge) in removed_edge:
continue
try:
backend_coupling_graph.remove_edge(edge[0], edge[1])
removed_edge.append(set(edge))
except NoEdgeBetweenNodes:
pass
return backend_coupling_graph
def get_zigzag_physical_layout(
num_orbitals: int, backend: BackendV2, score_layouts: bool = True
) -> tuple[list[int], int]:
"""The main function that generates the zigzag pattern with physical qubits that can be used
as an `intial_layout` in a preset passmanager/transpiler.
Args:
num_orbitals (int): Number of orbitals.
backend (BackendV2): A backend.
score_layouts (bool): Optional. If `True`, it uses the `lightweight_layout_error_scoring`
function to score the isomorphic layouts and returns the layout with less erroneous qubits.
If `False`, returns the first isomorphic subgraph.
Returns:
A tuple of device compliant layout (list[int]) with zigzag pattern and an int representing
number of alpha-beta-interactions.
"""
backend_coupling_graph = _make_backend_cmap_pygraph(backend=backend)
G, num_alpha_beta_qubits = create_lucj_zigzag_layout(
num_orbitals=num_orbitals,
backend_coupling_graph=backend_coupling_graph,
)
isomorphic_mappings = rustworkx.vf2_mapping(
backend_coupling_graph, G, subgraph=True
)
isomorphic_mappings = list(isomorphic_mappings)
edges = list(G.edge_list())
layouts = []
for mapping in isomorphic_mappings:
initial_layout = [None] * (2 * num_orbitals + num_alpha_beta_qubits)
for key, value in mapping.items():
initial_layout[value] = key
layouts.append(initial_layout)
two_q_gate_name = IBM_TWO_Q_GATES.intersection(
backend.configuration().basis_gates
).pop()
if score_layouts:
scores = lightweight_layout_error_scoring(
backend=backend,
virtual_edges=edges,
physical_layouts=layouts,
two_q_gate_name=two_q_gate_name,
)
return scores[0][0][:-num_alpha_beta_qubits], num_alpha_beta_qubits
return layouts[0][:-num_alpha_beta_qubits], num_alpha_beta_qubits
initial_layout, _ = get_zigzag_physical_layout(num_orbitals, backend=backend)
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend, initial_layout=initial_layout
)
# without PRE_INIT passes
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/o pre-init passes): {isa_circuit.count_ops()}")
# with PRE_INIT passes
# We will use the circuit generated by this pass manager for hardware execution
pass_manager.pre_init = ffsim.qiskit.PRE_INIT
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/ pre-init passes): {isa_circuit.count_ops()}")
Gate counts (w/o pre-init passes): OrderedDict({'rz': 12438, 'sx': 12169, 'cz': 3986, 'x': 573, 'measure': 52, 'barrier': 1})
Gate counts (w/ pre-init passes): OrderedDict({'sx': 7059, 'rz': 6962, 'cz': 1876, 'measure': 52, 'x': 35, 'barrier': 1})
Schritt 3: Utföhren mit Qiskit-Primitiven
Na de Optimierung vun den Schaltkreis för de Hardware-Utföhren sünd wi praat, em op de Teel-Hardware uttof öhren un Stichproben för de Grundtostandsenergieschattung to sammeln. Do wi blots een Schaltkreis hebbt, bruukt wi den Job-Utföhrenmodus vun Qiskit Runtime un föhrt uns Schaltkreis ut.
sampler = Sampler(mode=backend)
job = sampler.run([isa_circuit], shots=100_000)
primitive_result = job.result()
pub_result = primitive_result[0]
Schritt 4: Naobearbeiten un Torüchgaav vun dat Ergebnis in dat wünschte klassische Format
Nu schatten wi de Grundtostandsenergie vun den Hamiltonian mit de Funktion diagonalize_fermionic_hamiltonian. Disse Funktion föhrt dat sülvkonsistente Konfigurationswedderherstellenverfahren ut, um de verrüschte Quantenstichproben iterativ to verfienern un de Energieschattung to verbetern. Wi geven een Callback-Funktion, dormit wi de Twischenergeb nisse för een späder Analyse spiekern künnt. Kiek in de API-Dokumentation för Verklarungen vun de Argumenten vun diagonalize_fermionic_hamiltonian.
from functools import partial
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
solve_sci_batch,
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
pub_result.data.meas,
samples_per_batch=samples_per_batch,
norb=num_orbitals,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
carryover_threshold=carryover_threshold,
callback=callback,
seed=12345,
)
Iteration 1
Subsample 0
Energy: -108.97781410104506
Subspace dimension: 28561
Subsample 1
Energy: -108.97781410104506
Subspace dimension: 28561
Subsample 2
Energy: -108.97781410104506
Subspace dimension: 28561
Iteration 2
Subsample 0
Energy: -109.05150860754739
Subspace dimension: 287296
Subsample 1
Energy: -109.08152283263908
Subspace dimension: 264196
Subsample 2
Energy: -109.11636893067873
Subspace dimension: 284089
Iteration 3
Subsample 0
Energy: -109.15878555367885
Subspace dimension: 429025
Subsample 1
Energy: -109.16462831275786
Subspace dimension: 473344
Subsample 2
Energy: -109.15895026995382
Subspace dimension: 435600
Iteration 4
Subsample 0
Energy: -109.17784051223317
Subspace dimension: 622521
Subsample 1
Energy: -109.1774651326829
Subspace dimension: 657721
Subsample 2
Energy: -109.18085212360191
Subspace dimension: 617796
Iteration 5
Subsample 0
Energy: -109.18636242518915
Subspace dimension: 815409
Subsample 1
Energy: -109.18451014767518
Subspace dimension: 837225
Subsample 2
Energy: -109.18333728638888
Subspace dimension: 857476
Visualisierung vun de Ergebnisse
De eerste Plot wiest, dat wi na een paar Iterationen de Grundtostandsenergie binnen ~41 mH schatten (chemische Genauigkeit warrt typisch as 1 kcal/mol 1.6 mH akzepteert). De Energie kann verbetert warrn, indeem mehr Iterationen vun de Konfigurationswedderherstellung erlööft warrn or de Antall vun de Stichproben pro Batch verhöcht warrt.
De tweede Plot wiest de dörchsnittliche Besettung vun jeeden Ruumorbital na de leste Iteration. Wi künnt seen, dat sowohl de Spin-Up- as ok de Spin-Down-Elektronen de eerste fief Orbitalen mit hoge Wahrschienlichkeit in uns Lösungen besettet.
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - exact_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()

Tutorial-Ümfraag
Maakt bi de korte Ümfraag mit, um Feedback to dit Tutorial to geven. Dien Insichten helpt uns, uns Inholtsanbeden un Brukerrfohren to verbetern.